
The Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte), BfArM, is the institution responsible for regulating medical devices in Germany. Headquartered in Bonn, BfArM operates under the Federal Ministry of Health.
BfArM also oversees the clinical investigations of medical devices in Germany. Clinical investigations are conducted to assess the safety and effectiveness of the medical device. They are only required for high risk devices or if the existing clinical data or clinical evaluation report for a product is inadequate.
The Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte), BfArM, is the institution responsible for regulating medical devices in Germany. Headquartered in Bonn, BfArM operates under the Federal Ministry of Health.
BfArM also oversees the clinical investigations of medical devices in Germany. Clinical investigations are conducted to assess the safety and effectiveness of the medical device. They are only required for high risk devices or if the existing clinical data or clinical evaluation report for a product is inadequate.

Medical Device Classification
Since Germany is a European Union member country, it follows the device classification system that of EU Medical Device Directives (MDD) soon to be replaced by the EU Medical Device Regulations (MDR). Medical device classification in Germany is in four risk-based classes: I, IIa, IIb, and III. Class I being the lowest risk while Class III being the highest risk.
Medical Device Registration Process
All medical devices registered within the European Union must obtain CE Mark. A CE mark is a widely accepted and known symbol which indicates that the device conforms to all regulations and requirements under the current European Union standards. The CE mark also allows for the device to be marketed freely within all EU member states. The objective of a CE mark is to save manufacturers time, cost, and effort in preparing and submitting onerous dossiers, separately in 28 different countries. With a CE mark, companies can market and sell their products simultaneously in all EU member states whether that is Germany, France or Croatia. However, several EU member countries require some additional requirements before the product can be marketed or sold in that country.
Note that in order to obtain a CE-mark, a foreign manufacturer first must appoint an Authorized Representative (AR), especially if they do not have any representation in the EU. The AR can be based in any of the EU member states. Their primary role is to act as the liaison between the Notified Body, Agency and the manufacturer. The AR would assist with the registration process and perform all postmarket surveillance activities.
Additionally, manufacturers must have a Quality Management System (QMS). The most widely accepted QMS is the ISO 13485 certification, which is valid for three years.
Germany is one of the countries in the EU that does require additional requirements outside of CE mark. BfArM has an electronic portal called DIMDI. Manufacturers have to complete the following tasks:
- If the manufacturer, importer or AR of the product is registered as a business in Germany, they must notify BfArM once the device is placed on the market, using the DIMDI system.
- Assign a Safety Officer and notify it on the same DIMDI system.
- All labeling and Instructions for Use must be in German.
- Adverse event reporting should be made to the respective institutes depending on the nature of the medical device.
Market overview
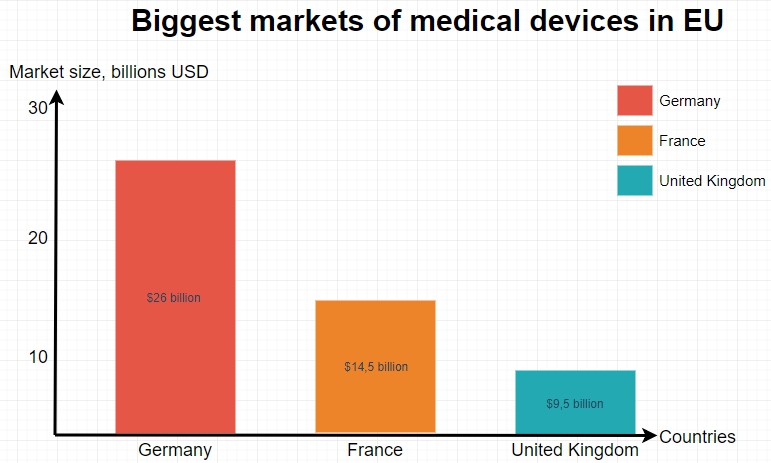
Germany has the largest medical device market in Europe and the third largest in the world. Germany’s most sizable competitors in terms of market size in Europe are the United Kingdom and France.
- Medical device market size in Germany: $26 billion USD
- Medical device market size in UK: $9.5 billion USD
- Medical device market size in France: $14.5 billion USD
Though Germany houses some of the biggest medical device companies in the world, it is dominated by small and medium-sized enterprises (SMEs). Germany is receptive to new and innovative technology to support their highly accredited medical care.
Products with the highest demand in Germany include cutting-edge technologies, diagnostic and therapeutic devices, and non-invasive or minimally invasive medical equipment.
Correct following of medical device regulations in Germany will help you to increase your positions on market.
About RegDesk
RegDesk(™) is a diverse platform that eliminates a need for medical device companies to spend months working to gather intelligence and prepare registration applications to bring their product to Germany, and any other country. We can make medical device registration process simple for you. By using RegDesk’s state-of-the-art platform, our clients will be able to receive expert answers to their regulatory questions, real-time alerts to regulatory changes happening in countries of their choice, and more! To get the full experience of the power of RegDesk, get in touch with any of our representatives.
